General Description
VERSE is designed for high-performance read summarization for next generation sequencing. VERSE is 50x faster than HTSeq when computing the same gene counts. It introduces a novel, hierarchical assignment scheme, which allows simultaneous quantification of multiple feature types or annotation levels without repeatedly assigning reads. There is also a set of parameters the user can use to fine-tune the assignment logic. VERSE can be readily incorporated into any existing RNA-Seq analysis pipelines.
Input file format: BAM/SAM produced by read alignment tools such as STAR, annotation file (GTF format).
Output files: Feature/Gene counts file with the same format as HTSeq/featureCounts, a summary file summarizing the results, read assignment details for each read/read pairs if user specifies -R.
Usage: ./verse [options] -a <annotation_file> -o <output_file> input_file
A sample command:
./verse -a testdata/test.gtf -t 'exon' -g gene_id -z 3 -s 1 -o testdata/intersection_nonempty.stranded.paired testdata/PE.sam
A sample hierarchical assign command:
./verse -a testdata/test.gtf -t 'exon;intron;xine' -g gene_id -z 3 -o testdata/intersection_nonempty.unstranded.paired.hierarchical testdata/PE.sam
Parameter Description
Required Parameters
-a <input> Give the name of the annotation file. The program currently only
supports GTF format.
-o <input> Give the general name of the output file, e.g., 'Sample_A'.
The summary file will be named 'Sample_A.summary.txt.'
The file containing gene counts will be named 'Sample_A.exon.txt',
'Sample_A.intron.txt', etc.
input_file Give the name of input read file that contains the read mapping
results. Format of input file is automatically determined (SAM/BAM)
If it is paired-end data, the file MUST be name-sorted, otherwise
the user MUST specify -S, or use samtools to sort it by name.
Optional Parameters:
-z <int> The Running Mode: 0 by default (featureCounts), 1 (HTSeq-Union),
2 (HTSeq-Intersection_strict), 3 (HTSeq-Intersection_nonempty),
4 (Union_strict), 5 (Cover_length).
Please refer to the manual or use '-Z' to check the details of
each mode.
-t <input> Specify the feature type. Only rows which have the matched
feature type in the provided GTF annotation file will be included
for read counting. 'exon' by default.
Providing a list of feature types (e.g., -t 'exon;intron;mito')
will automatically enter hierarchical_assign mode. If the user
wants to assign independently for each feature type, please
specify '--assignIndependently'. Use -Z to check the details.
-g <input> Specify the gene_identifier attribute, which is normally 'gene_id'
or 'gene_name'. 'gene_id' by default.
-S If the input file is paired-end data but not sorted by read name,
this option MUST be specified.
-s <int> Indicate if strand-specific read counting should be performed.
It has three possible values: 0 (unstranded), 1 (stranded) and
2 (reversely stranded). 0 by default.
-Q <int> The minimum mapping quality score a read must satisfy in order
to be counted. For paired-end reads, at least one end should
satisfy this criteria. 0 by default.
-l Output the gene length. This length is feature_type specific,
which means the length in an exon_count file will be the total
exon length of each gene, the length in an intron_count file will
be the total intron length, etc.
-R Output read assignment details for each read/read pairs. The
details will be saved to a tab-delimited file that contains five
columns including read name, status(assigned or the reason if not
assigned), feature type and assigned gene name.
-T <int> Number of threads used for read assignment. 1 by default.
Note that when running, VERSE will initiate one main thread and
at least one helper thread for read assignment. This option
specifies the number of helper threads.
--singleEnd If specified, VERSE will assume the input is
single-end data, and assign every reads individually. If this is
not specified(default), the input will be treated as paired-end
data. The 'missing mate' count will show how many reads are not
in pairs. VERSE can correctly assign single-end data in paired-
end mode, but it will take longer and the reads will all be
counted as missing mates. So it is recommended to specify this
if the user knows it is single-end.
--assignIndependently If specified, VERSE will assign reads to listed
feature types independently. This has the same effect as running
VERSE separately for each feature type, except that it only
requires one run, thus is more efficient.
--readExtension5 <int> Reads are extended upstream by <int> bases from
their 5' end.
--readExtension3 <int> Reads are extended upstream by <int> bases from
their 3' end.
--minReadOverlap <int> Specify the minimum number of overlapped bases
required to assign a read to a feature. 1 by default.
--maxReadNonoverlap <int> Specify the maximum number of non-overlapped bases
a read can have. A read is considered no_feature if its number of
non-overlapped bases exceeds this threshold. This option is not
useful in strict mode since it requires the assigned feature to
overlap every base of the read.
--countSplitAlignmentsOnly If specified, only split alignments (CIGAR
strings containing letter `N') will be counted. All the other
alignments will be ignored. An example of split alignments is
the exon-spanning reads in RNA-seq data.
--read2pos <5:3> The read is reduced to its 5' most base or 3'
most base. Read summarization is then performed based on the
single base which the read is reduced to.
--ignoreDup If specified, reads that were marked as
duplicates will be ignored. Bit Ox400 in FLAG field of SAM/BAM
file is used for identifying duplicate reads. In paired end
data, the entire read pair will be ignored if at least one end
is found to be a duplicate read.
--minDifAmbiguous <int> This option can only be used in VERSE-cover_length
mode. When the read or the read pair hits more than one genes,
the number of bases overlapped by each gene will be calculated.
If the covered_length difference between the most covered gene
and the second most covered gene is smaller than this specified
value, the read will be set ambiguous. 1 base difference by default.
--nonemptyModified This option can only be used in intersection_
nonempty mode. In cases where HTSeq would assign multi-hit reads
to no_feature, VERSE will assign those to ambiguous.
--multithreadDecompress BAM files generated with SAMTools or STAR after
year 2013 has a slight format change which allows multithreaded
decompression. BAM processing will be faster if this option is
specifed and multiple core is used.
Optional Paired-end Parameters:
-P If specified, template length (TLEN in SAM specification) will be
checked when assigning read pairs (templates) to genes. This option
is only applicable in paired-end mode. The distance thresholds
should be specified using -d and -D options.
-d <int> Minimum template(read pair) length, 50 by default.
-D <int> Maximum template(read pair) length, 600 by default.
-B If specified, only read pairs that have both ends successfully
aligned will be considered for summarization. This option is only
applicable for paired-end reads.
-C If specified, the chimeric read pairs (those that have their two
ends aligned to different chromosomes) will NOT be included for
summarization. This option is only applicable for paired-end data.
Additional Information:
-v Output version of the program.
-Z Show details about the running mode or scheme.
Running mode parameters
The main differences between modes are the stringency of assignment and the method of assigning reads that overlap multiple features.
featureCounts Mode:
-z 0 featureCounts(default): Quantify by overlapping and voting.
If the read pair overlaps multiple genes, it will assign the read pair
to the gene that is overlapped by both reads.
Please refer to the SUBREAD users guide for more information.
This mode is developed by Drs. Yang Liao, Gordon K Smyth and Wei Shi.
http://bioinf.wehi.edu.au/featureCounts.
HTseq Modes:
-z 1 HTSeq-Union: Quantify by overlapping.
If the read (or read pair) overlaps multiple genes, it will be set
ambiguous. Only reads that overlap one gene will be assigned.
-z 2 HTSeq-Intersection_strict: Quantify by overlapping and intersection.
This mode requires the assigned gene to cover every base of the read.
If more than one such genes exist, the read is set ambiguous.
-z 3 HTSeq-Intersection_nonempty: Quantify by overlapping and intersection.
This mode does NOT require the assigned gene to cover every base of
the read, but the gene must cover all sections of the read that overlap
genes. If more than one such genes exist, the read is set ambiguous.
Please refer to the HTSeq documentation for more information.
HTSeq is developed by Dr. Simon Anders at EMBL Heidelberg.
http://www-huber.embl.de/HTSeq/doc/count.html#count.
The actual implementation of the HTseq scheme is different in VERSE.
VERSE Modes:
-z 4 Union_strict: A combination of HTSeq-Union and HTSeq-Intersection_strict.
This mode requires every base of the read to overlap one and only one gene.
This mode is the most conservative.
-z 5 Cover_length: Quantify by overlapping length comparison.
After getting a list of overlapping genes, VERSE will calculate the
overlapping length of each gene and assign the read to the most covered
gene. One can use --minDifAmbiguous to set the minimum allowed coverage
difference between the most covered gene and the gene with the second
highest coverage. If the difference is not large enough the read will
be set ambiguous.
Assignment Scheme Parameters
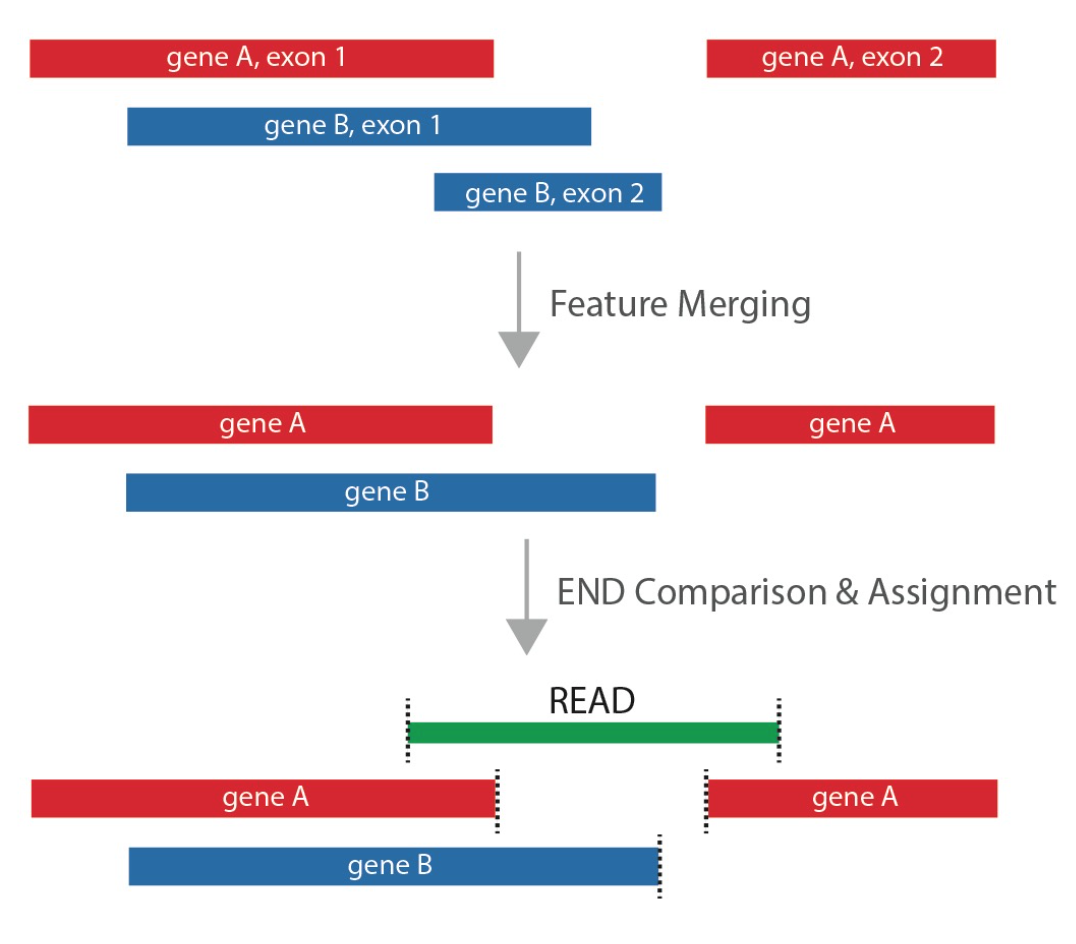
When assigning reads to different feature types, e.g., exons, introns, antisense exons, antisense introns, intergenic regions, mitochondria, etc., or level 1, level 2, level 3 transcripts in the GENCODE annotation, VERSE is capable of assigning reads to all the feature types within a single run, provided a combined annotation GTF file with all the feature types.
Hierarchical_assign:
VERSE offers a hierarchical_assign scheme, which ensures that each read
is assigned only once to the most interested feature type. When user
inputs a list of feature types, e.g., 'exon;intron;mito', VERSE will
assume the first feature type (exon) in the list is of the highest priority,
and will try assigning reads to it first, using the specified mode (e.g.,
union mode). The reads that cannot be assigned to exon will enter the
next round of assignment to the next feature type, i.e., intron.
This procedure will repeat until every read is successfully assigned,
or fails to be assigned to any of the feature types.
Independent_assign:
If '--assignIndependently' is specified, instead of running hierarchical
_assign, VERSE will assign reads to feature types independently.
This has the same effect as running VERSE separately for each feature type,
except that it only requires one run, thus is more efficient.
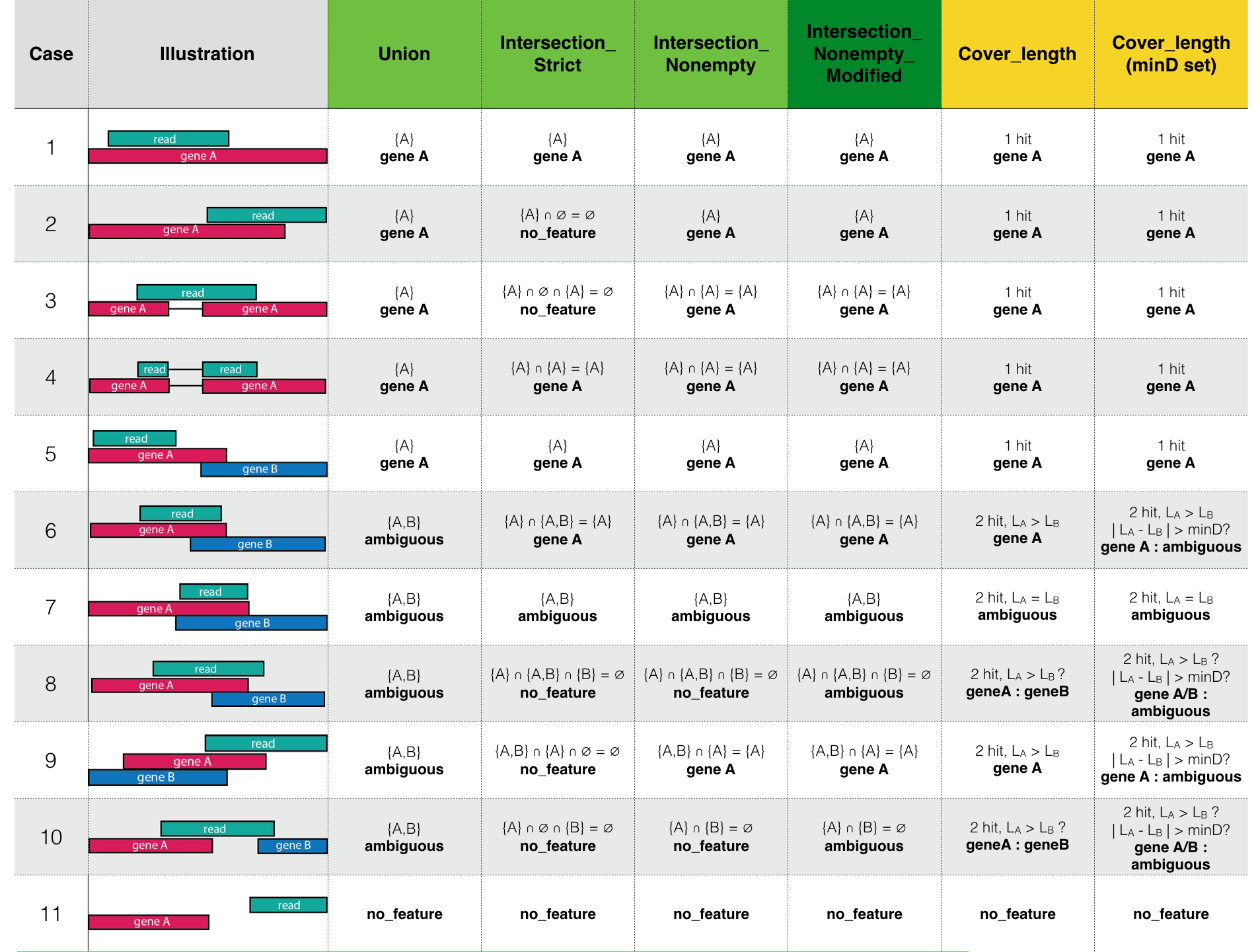
Running Mode Details
This table illustrates the difference between the modes implemented in VERSE.
Green: HTSeq modes, based on intersection.
Yellow: VERSE modes, based on length.
Note that featureCounts mode is not illustrated here. When quantifying in single-end mode, featureCounts behaves the same as the union-mode of HTSeq, discarding reads overlapping multiple exons. In paired end mode, it yields results similar to intersection-nonempty because it preferentially assigns fragments to genes that hit both reads.
(Click on images for larger version)
Note the Intersection_Nonempty_Modified mode is different from the original HTSeq mode in Case 8 and 10, where it assigns the read to ambiguous rather than to no_feature. This makes more sense, especially when assigning the reads hierarchically to different feature types.
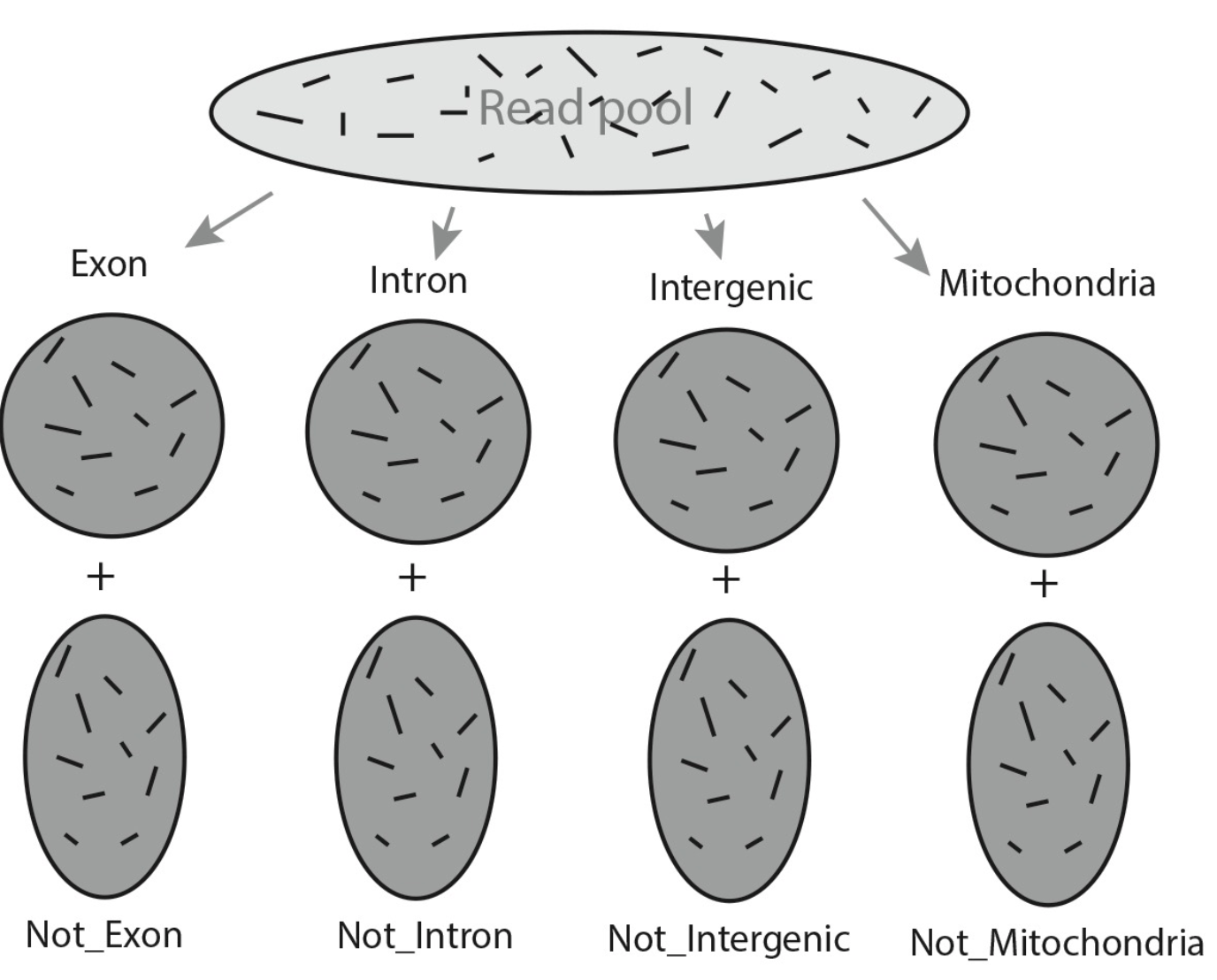
Assigning Reads to Multiple Feature Types
Traditional methods require multiple separate runs to assign reads to different feature types. VERSE allows all reads to be assigned to all feature types within a single run. The independent assign scheme produce the same results as traditional methods, while hierarchical scheme assign reads sequentially to each feature type according to their priority, as illustrated in the figures below.
To use multiple feature type assignment, VERSE require the input annotation file to be a combined annotation with all feature types included.
Independent Assign
Assigned Exon + Assigned Intron + Assigned Intergenic + Assigned Mitochondria + Ambiguous + No_feature ≠ Total # of reads
Hierarchical Assign
Assigned Exon + Assigned Intron + Assigned Intergenic + Assigned Mitochondria + Ambiguous + No_feature = Total # of reads
Feature type at the first level (e.g., exon) will obtain the same gene counts as the independent assign scheme. Feature type at lower level (e.g., intron) in the hierarchy will get less counts, but since reads assigned to exons has already been removed, quantification at lower level will get less false positives.
The design of the hierarchy thus is extremely important and should address the main research interest.