VERSE
| Maintained by Qin Zhu | License (GNU) |
A versatile and efficient RNA-Seq read counting tool
VERSE is designed for high-performance read summarization for next generation sequencing. VERSE is 50x faster than HTSeq when computing the same gene counts. It introduces a novel, hierarchical assignment scheme, which allows simultaneous quantification of multiple feature types or annotation levels without repeatedly assigning reads. There is also a set of parameters the user can use to fine-tune the assignment logic. VERSE can be readily incorporated into any existing RNA-Seq analysis pipelines.
VERSE is implemented in C. It is built on top of featureCounts. VERSE supports Mac OSX and linux systems.
Publication: Zhu, Q., Fisher, S.A., Shallcross, J., Kim, J. (Preprint). VERSE: a versatile and efficient RNA-Seq read counting tool. bioRxiv 053306.
doi: http://dx.doi.org/10.1101/053306
Feature Summary
- VERSE supports the following modes of RNA-Seq quantification:
- FeatureCounts (Default)
- HTSeq Union (-z 1)
- HTSeq Intersection-strict (-z 2)
- HTSeq Intersection-nonempty (-z 3)
- VERSE Union-strict (-z 4)
- VERSE Cover-length (-z 5)
- Supported Quantification Schemes:
- Hierarchical Assign -- assign reads to feature types according to their priority.
- Independent Assign -- assign reads to feature types independently in a single run.
Update Log
- v1.0.5 - Bug Fixed: Corrected counting for Non-unique mapped reads. New Feature: --multithreadDecompress. (9/10/2015)
- v1.0.4 - Bug Fixed: Fixed crash cases where there are no unprocessed reads but thread was initiated; fixed merging of WASH6P. New Feature: --nonemptyModified. (7/14/2015)
- v1.0.3 - Bug Fixed: Only one read in a BGZF chunk situation. (6/30/2015)
Download
- verse_0.1.5.zip - the latest stable release.
Installation:
-
cdto the src folder, usemaketo compile the code.For Linux OS, use command::
make -f Makefile.Linux
For Mac OS, use command:
make -f Makefile.MacOS
Usage
-
Please run
./verseto see the details.A sample command:
./verse -a testdata/test.gtf -t 'exon' -g gene_id -z 3 -s 1 -o testdata/intersection_nonempty.stranded.paired testdata/PE.sam
A sample hierarchical assign command:
./verse -a testdata/test.gtf -t 'exon;intron;xine' -g gene_id -z 3 -o testdata/intersection_nonempty.unstranded.paired.hierarchical testdata/PE.sam
- User Manual (html)
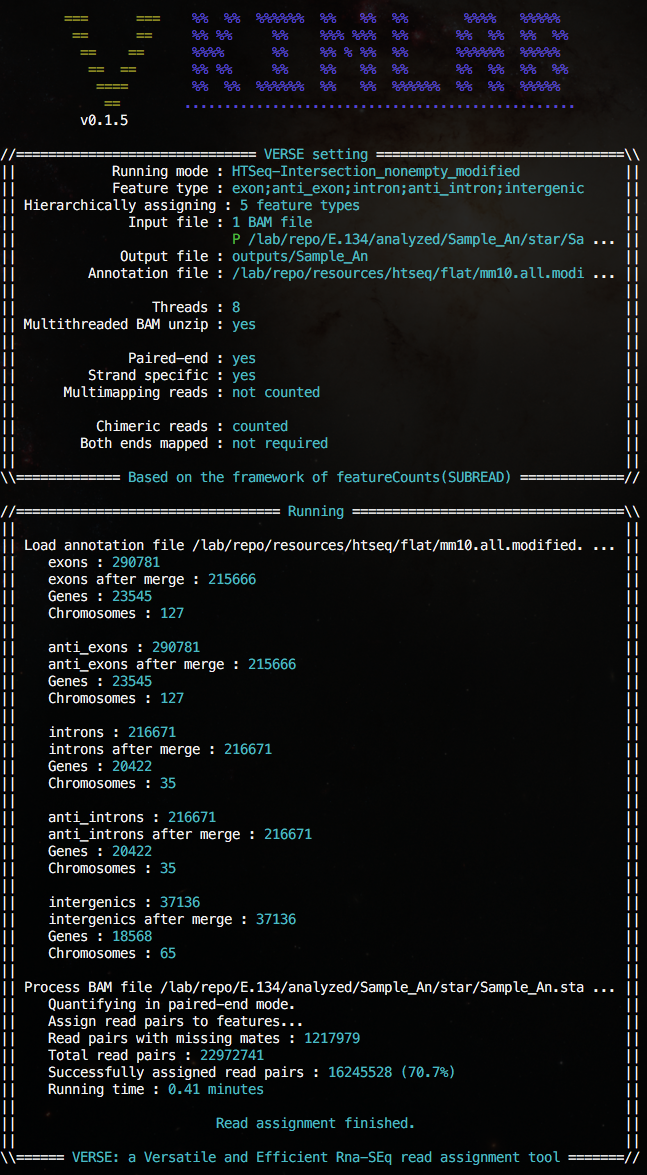
Screen shots
(Click on images for larger version)A sample hierarchical assignment which assigns all reads to exon, antisense exon, intron, antisense intron and intergenic regions sequentiallly in a single run.