NoFold: RNA structure clustering without folding or alignment
| Maintained by Sarah Middleton | License (pdf) |
Publication: Middleton, S.A. and Kim, J. 2014. NoFold: RNA structure clustering without folding or alignment. RNA 20: 1671-1683.
About NoFold
NoFold is an approach for characterizing and clustering RNA secondary structures without computational folding or alignment. It works by mapping each RNA sequence of interest to a structural feature space, where each coordinate within the space corresponds to the probabilistic similarity of the sequence to an empirically defined structure model (e.g. Rfam family covariance models). NoFold provides scripts for mapping sequences to this structure space, extracting any robust clusters that are formed, and annotating those clusters with structural and functional information.
Recent updates
10-03-16 - Latest nightly source (21 MB)02-26-14 - The paper version of NoFold (1.0.1) has been added.
02-25-14 - Supplemental data and results files for the paper have been updated.
Download for Linux
- NoFold 1.0.1 (19 MB) - latest stable release
- README (17 KB)
Included in download:
- All code needed to run NoFold (scoring, clustering, annotation)
- 1,973 calibrated Rfam covariance models
- Pre-made threshold files appropriate for datasets of up to ~4,000 sequences
- A script for generating thresholds specific to your dataset size, if needed
- A demo dataset for testing your installation
- Python 2.X - Used to run the most of the code
- Infernal (v.1.0.2) (infernal-1.0.2.tar.gz, 15.2 MB) - Used for scoring
- R with fastcluster package - Used for clustering and some statistics
- LocARNA - Used to predict consensus structure of clusters
- RNAz (optional) - used to get additional stats on cluster structures
Git repository
Getting started
- Install required software. Add executables to your PATH if possible, otherwise you will need to supply a path to the folders containing the executables to NoFold (see README).
- Unzip NoFold: tar -zxvf nofold.tar.gz
- Navigate to /src/ directory: cd nofold/src
- Test that everything is working by running the demo dataset. See README for instructions.
Example usage
Using demo1.db (included with NoFold):
python score_and_normalize.py ../demo/demo1/demo1.db --cpus=4 python nofold_pipeline.py ../demo/demo1/demo1.zNorm.pcNorm100.zNorm.bitscore ../demo/demo1/demo1.db \ --cpus=4 --bounds-file=../thresh/bounds_30seq.txt --verbose
This scores the sequences and then extracts clusters based on the within-cluster distance thresholds defined in the bounds file. It outputs a file with annotation information about each identified cluster (example).
Paper data
Supplemental files:
- Supplemental analyses: pdf (361 KB)
- RESS axes loadings: txt (1.8 MB)
- RESS axes correlations: txt (246 KB)
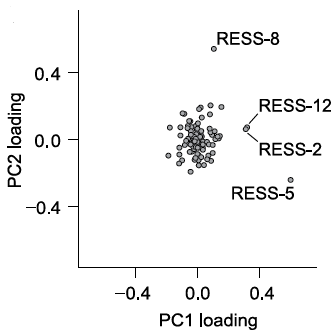
- RESS axes loadings for synthetic structure PCA (Fig. 2B): png (12 KB)
- Rfam test set LDA (after CM removal): txt (716 KB)
- Rfam test set - distribution of sequences per cluster: pdf
- Rfam test set - distribution of cluster diameters: pdf
- Experimental datasets - distribution of sequences per cluster: pdf
- Experimental datasets - distribution of cluster diameters: pdf
{kind=link}
Datasets & clustering results
- Synthetic structure test set
- Plain sequences: sequences / clusters / all data zip (3 MB)
- Rfam 20-family test set
- Plain sequences: sequences / clusters / all data zip (34 MB)
- Embedded sequences: sequences / clusters / all data zip (39 MB)
- Plain seqs + bg: sequences / clusters / all data zip (184 MB)
- Full Rfam "cross validation" set
- Plain sequences: all data zip (989 MB)
- Result summary: xlsx (183 KB)
- Dendritically localized transcripts, 3' UTRs
- 50nt window: sequences / clusters / all data zip (143 MB)
- 150nt window: sequences / clusters / all data zip (22 MB)
- Dendritically localized transcripts, retained introns
- 50nt window: sequences / clusters / all data zip (45 MB)
- 150nt window: sequences / clusters / all data zip (13 MB)
- Non-canonical translation init sites
- 50nt window: sequences / clusters / all data zip (14 MB)
- 150nt window: sequences / clusters / all data zip (12 MB)
- All paper data (510 MB)